The Trending With Impact series highlights Aging (Aging-US) publications that attract higher visibility among readers around the world online, in the news, and on social media—beyond normal readership levels. Look for future science news about the latest trending publications here, and at Aging-US.com.
—
In 1961, Leonard Hayflick and Paul Moorhead proposed a theory later named the Hayflick Limit. They discovered that a normal human cell can divide between 50 and 70 times before it can no longer proliferate and eventually dies. Researchers have since continued to explore this phenomenon and, today, this aging process is known as cellular (replicative) senescence.
“There are currently several experimental models of cellular senescence. Hayflick and Moorhead observed that primary human fibroblasts in culture exhibit a limited proliferative capacity [6]. This growth arrest during passages is called replicative senescence.”
This permanent cessation of the cell cycle is universally found in biology due to known and unknown causes, including the shortening of telomeres. While telomere shortening plays an important role, it is not the only event responsible for inducing cellular senescence. Thus, researchers have spent decades under the microscope experimenting with cellular models of replicative senescence.
In a new study released on April 4, 2022, researchers from Sapporo Medical University in Sapporo, Japan, investigated mechanisms of replicative senescence in vitro. Their research paper was published on the cover of Aging (Aging-US) Volume 14, Issue 7, and entitled, “Downregulation of IGFBP5 contributes to replicative senescence via ERK2 activation in mouse embryonic fibroblasts.”
The Study
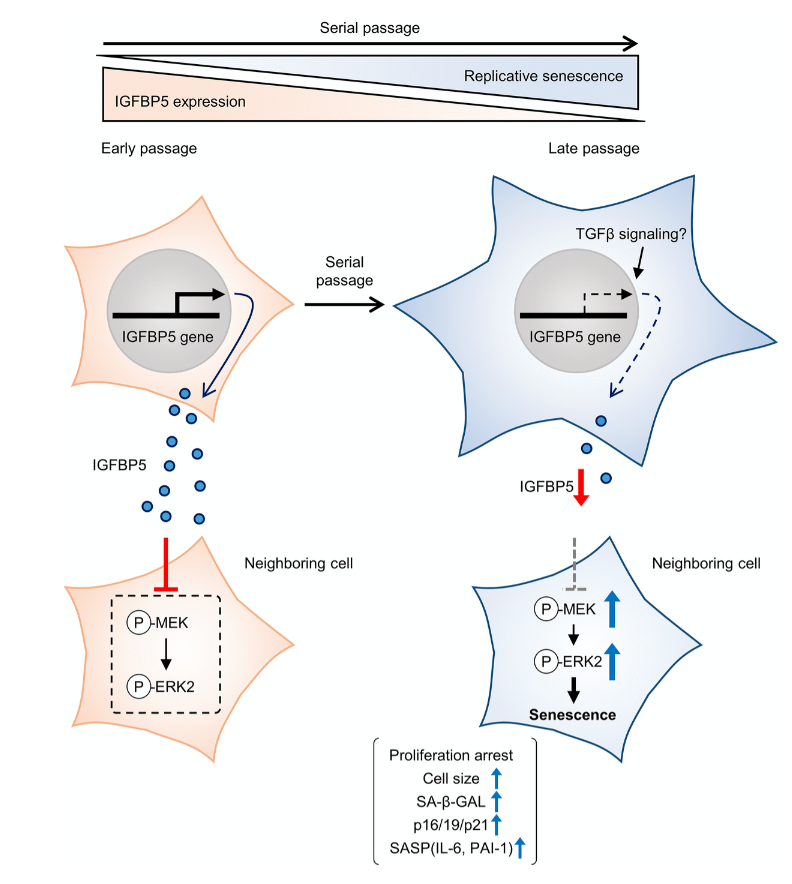
Cellular senescence is typically characterized by cell growth arrest, an increase of cells positive for SA-β -gal staining, and upregulation of p16 and p19. To begin this study, the team cultured embryonic mouse fibroblasts (MEFs) and conducted cell passages according to the 3T3 method. They found that the MEFs underwent senescence after the 5th passage (P5). The team also found that at P8, the expression of insulin-like growth factor binding protein 5 (IGFBP5) mRNA was significantly reduced when compared with that of P2 MEFs.
Next, the team performed a knockdown of IGFBP5 in the MEF cells. Results showed that IGFBP5 knockdown induced premature cellular senescence in P2 MEFs. Knockdown of IGFBP5 increased phosphorylation of extracellular signal-regulated kinases 1 and 2 (ERK1/2) but did not affect expression levels of Akt or p16 repressors. The researchers also found that supplementing the cell culture growth medium with additional exogenous IGFBP5 delayed growth arrest and reduced replicative senescence in the MEF cells.
“To examine whether activated ERK1 and ERK2 by IGFBP5 knockdown are involved in the induction of senescent phenotypes, we examined effects of knockdown of ERK1 and ERK2 using a combination with IGFBP5 siRNA in P2 MEFs.”
Upon further analysis of ERK1/2’s role in IGFBP5-knockdown cells, the team found that the silencing of ERK2, and not ERK1, blocked the increase in the number of SA-β-GAL-positive cells. ERK2 knockdown attenuated the reduction in the cell number and upregulation of p16 and p21 in IGFBP5-knockdown cells. This study provides evidence that downregulation of IGFBP5 contributes to replicative senescence via ERK2 activation in mouse embryonic fibroblasts.
Conclusion
For the first time, the role of IGFBP5 in replicative senescence was demonstrated in MEFs. Their findings suggest that ERK2 underlies cellular senescence induced by IGFBP5 downregulation. Cellular senescence appears to be a complex process with many moving parts. While more research is needed to fully understand the role of IGFBP5 in replicative senescence, this study provides new insights into the underlying mechanisms involved in this complex process.
“In conclusion, the results of the present study demonstrated that downregulation of IGFBP5 during serial passage contributes to replicative senescence via an ERK2-dependent mechanism (Figure 6). The results suggest that IGFBP5 counteracts replicative senescence in MEFs.”
Click here to read the full research paper published by Aging (Aging-US).
AGING (AGING-US) VIDEOS: YouTube | LabTube | Aging-US.com
—
Aging (Aging-US) is an open-access journal that publishes research papers bi-monthly in all fields of aging research. These papers are available at no cost to readers on Aging-us.com. Open-access journals have the power to benefit humanity from the inside out by rapidly disseminating information that may be freely shared with researchers, colleagues, family, and friends around the world.
For media inquiries, please contact [email protected].